Pathway example¶
Lets start with a pathway downloaded from the KEGG database.
The Python code in this example is available as a script.
First lets define a parsing function for the KEGG formatted pathways file.
def get_genes(pathway):
fid = open(os.path.join(".",pathway+".txt"),'r')
isGene = False
gene2symbol = {}
for linja in fid:
linja = linja[:-1]
if re.search("^GENE",linja):
isGene = True
elif re.search("^[A-Z]+",linja):
isGene = False
if isGene:
geneIds = linja.split(";")[0]
_geneIds = [re.sub("\s+","",gi) for gi in geneIds.split(" ")]
geneIds = []
for gid in _geneIds:
if len(gid) > 0 and gid != 'GENE':
geneIds.append(gid)
if len(geneIds) != 2:
print("...%s"%str(geneIds))
raise Exception("Could not parse gene names")
ncbiId,symbol = geneIds
gene2symbol[ncbiId] = symbol
fid.close()
return gene2symbol
Extract the genes into a list
>>> pathwayFile = "hsa00860.txt"
>>> pathway = pathwayFile[:-4]
>>> geneList = get_genes(pathway)
>>> print(geneList.items()[:3])
[('7363', 'UGT2B4'), ('212', 'ALAS2'), ('210', 'ALAD')]
Specify a location for the analysis files.
>>> gsaDir = os.path.join(".","gsa-path")
>>> if not os.path.exists(gsaDir):
>>> os.mkdir(gsaDir)
Make imports and specify variables
>>> from htsint import GeneOntology,TermDistances,GeneDistances
>>> useIea = True
>>> aspect = "biological_process"
>>> _aspect = 'bp'
>>> taxaList = ['9606']
>>> go = GeneOntology(taxaList,useIea=useIea,aspect=aspect)
>>> termsPath = os.path.join(gsaDir,"go-terms-%s.pickle"%(_aspect))
>>> graphPath = os.path.join(gsaDir,"go-graph-%s.pickle"%(_aspect))
By default htsint works at the genome level so to get term distances with respect to a subset of genes we need to specify an accepted list. The rest of the process is the same as was shown in the gsa example.
Create the gene-term dictionaries
>>> geneIds = geneList.keys()
>>> go.create_dicts(termsPath,accepted=geneIds)
>>> gene2go,go2gene = go.load_dicts(termsPath)
>>> print("pathway genes with terms: %s/%s"%(len(gene2go.keys()),len(geneIds)))
Create the term specific graph
>>> G = go.create_gograph(termsPath=termsPath,graphPath=graphPath)
>>> print("Term graph for with %s nodes successfully created."%(len(G.nodes())))
Calculate the distances between terms
>>> termDistancePath = os.path.join(gsaDir,"term-distances-%s.npy"%(_aspect))
>>> td = TermDistances(termsPath,graphPath)
>>> print("total distances to evaluate: %s"%td.totalDistances)
>>> td.run_with_multiprocessing(termDistancePath,cpus=4)
Map the term distances into gene space
>>> geneDistancePath = os.path.join(gsaDir,"gene-distances-%s.csv"%(_aspect))
>>> gd = GeneDistances(termsPath,termDistancePath,outFile=geneDistancePath)
>>> gd.run()
Run the parameter search for spectral clustering
>>> silvalFile = re.sub("\.csv","-scparams-sv.csv",geneDistancePath)
>>> clustersFile = re.sub("\.csv","-scparams-cl.csv",geneDistancePath)
>>> scps = SpectralClusterParamSearch(geneDistancePath,dtype='distance')
>>> scps.run(chunks=5,kRange=range(3,11))
Plot the parameter search
>>> psFigureFile = os.path.join(gsaDir,"param-scan-%s.png"%(_aspect))
>>> scr = SpectralClusterResults(silvalFile,clustersFile)
>>> scr.plot(figName=psFigureFile)
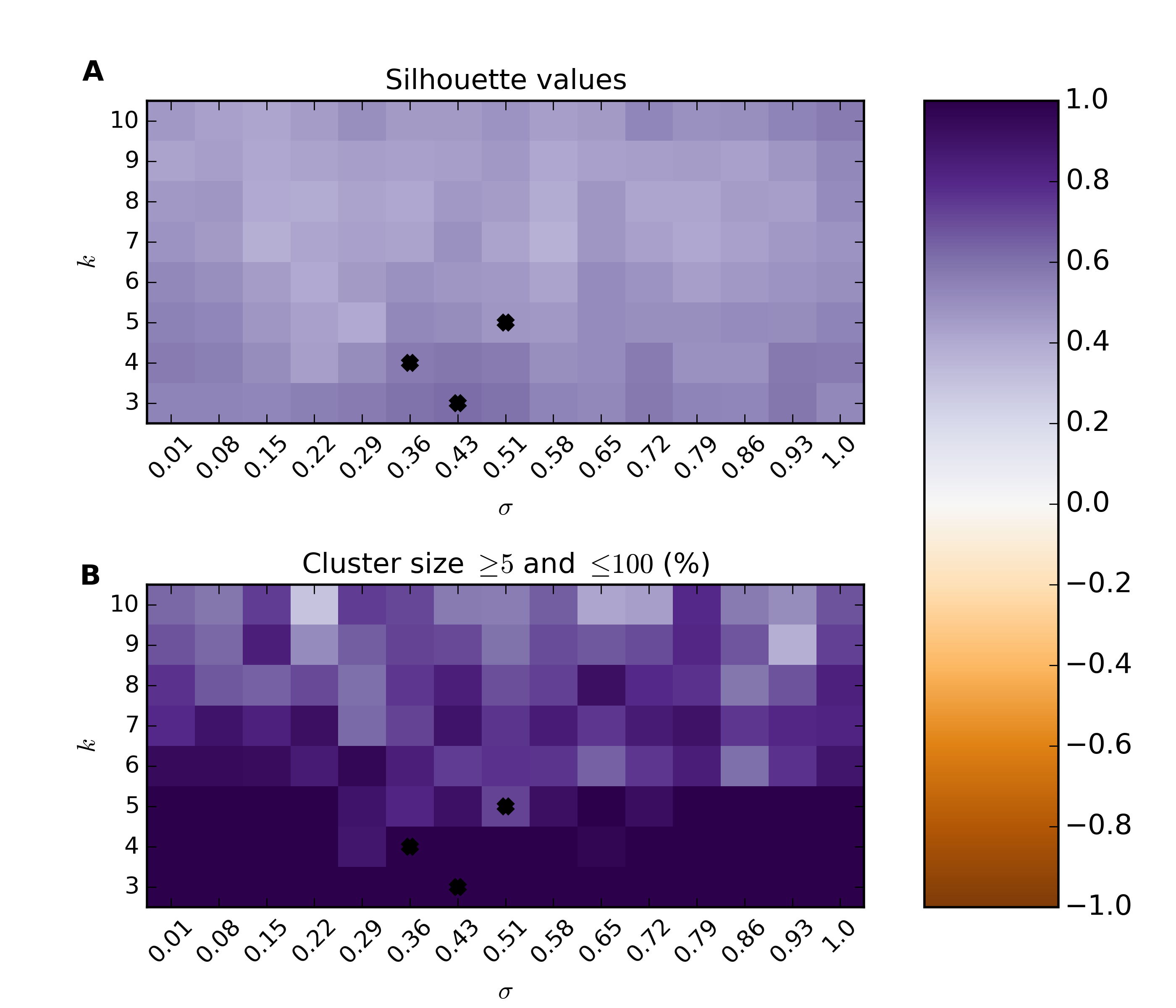
Run spectral clustering with selected parameters
>>> k = 3
>>> sigma = 0.43
>>> labelsPath = os.path.join(gsaDir,"sc-labels-%s.csv"%(_aspect))
>>> sc = SpectralCluster(geneDistancePath,dtype='distance')
>>> sc.run(k,sk=None,sigma=sigma,verbose=True)
>>> sc.save(labelsPath=labelsPath)
The genes and their labels are saved in labelsPath.