Assembly summary and statistics¶
We carried out the de novo transcriptome assembly with the software pacakge Trinity [Grabherr11]. The Trinity software suite consists of three main pieces:
- Inchworm - assembles the RNA-seq data into the unique sequences of transcripts, often generating full-length transcripts for a dominant isoform, but then reports just the unique portions of alternative ly spliced transcripts.
- Chrysalis - clusters the Inchworm contigs into clusters and constructs complete de Bruijn graphs for each cluster. Each cluster represents the full transcriptonal complexity for a given gene (or sets of genes that share sequences in common). Chrysalis then partitions the full read set among these disjoint graphs.
- Butterfly then processes the individual graphs in parallel, tracing the paths that reads and pairs of reads take within the graph, ultimately reporting full-length transcripts for alternatively spliced isoforms, and teasing apart transcripts that corresponds to paralogous genes.
Sequence preprocessing¶
A Python script was used to unzip, concatenate and trim the original reads via system calls (subprocess). We used Trimmomatic [Bolger14] to trim reads based on quality with default settings (LEADING:5 TRAILING:5 MINLEN:36).
There are four output files. Two are for the paired output where both reads passed, and two are for corresponding unpaired output where only one read passed (see manual).
Running Trinity¶
A Python script was used to generate the Trinity arguments.
The generalized Trinity command is shown below.
~$ export TRINITY_HOME="/usr/src/trinityrnaseq-2.0.4" ~$ $TRINITY_HOME/Trinity --seqType fq --output /path/to/out --trimmomatic --full_cleanup --SS_lib_type FR --max_memory 26G --CPU 29 --normalize_reads --left left1.fastq,left2.fastq,left3.fastq --right right1.fastq,right2.fastq,right3.fastq 2>&1 | tee ./run-trinity-gg.log
This produces the output file ./<TRINITY_OUT>/Trinity.fasta which we can run some basic statistics on.
Transcriptome summary¶
Trinity groups transcripts into clusters that are loosely referred to as a gene. The accession identifiers in the ./trinity_out_dir/Trinity.fasta encode gene and isoform information. Per the documentation (see links below) if we have the accession >c0_g1_i1 this refers to Trinity read cluster c0, gene g1 and isoform i1. The gene identifier in this case is c0_g1.
The basic stats of the assembly are:
~$ export TRINITY_HOME="/usr/src/trinityrnaseq-2.0.4" ~$ $TRINITY_HOME/util/TrinityStats.pl ~/sequencing/pieris/dn-trinity/Trinity.fasta ################################ ## Counts of transcripts, etc. ################################ Total trinity 'genes':65012 Total trinity transcripts:98416 Percent GC: 38.04 ######################################## Stats based on ALL transcript contigs: ######################################## Contig N10: 4650 Contig N20: 3779 Contig N30: 3184 Contig N40: 2645 Contig N50: 2155 Median contig length: 595 Average contig: 1151.58 Total assembled bases: 113334017 ##################################################### ## Stats based on ONLY LONGEST ISOFORM per 'GENE': ##################################################### Contig N10: 4325 Contig N20: 3444 Contig N30: 2704 Contig N40: 2056 Contig N50: 1496 Median contig length: 403 Average contig: 807.01 Total assembled bases: 52465576
Map the reads using BLAST¶
- First, we BLAST the transcript against SwissProt (-c can be used to initiate cluster mode)
- Then, these data were are parsed and summarized.
For each transcript the top BLAST match with the best score was kept. The minimum BLAST e-value was set at 0.0001.
SwissProt (isoforms)¶
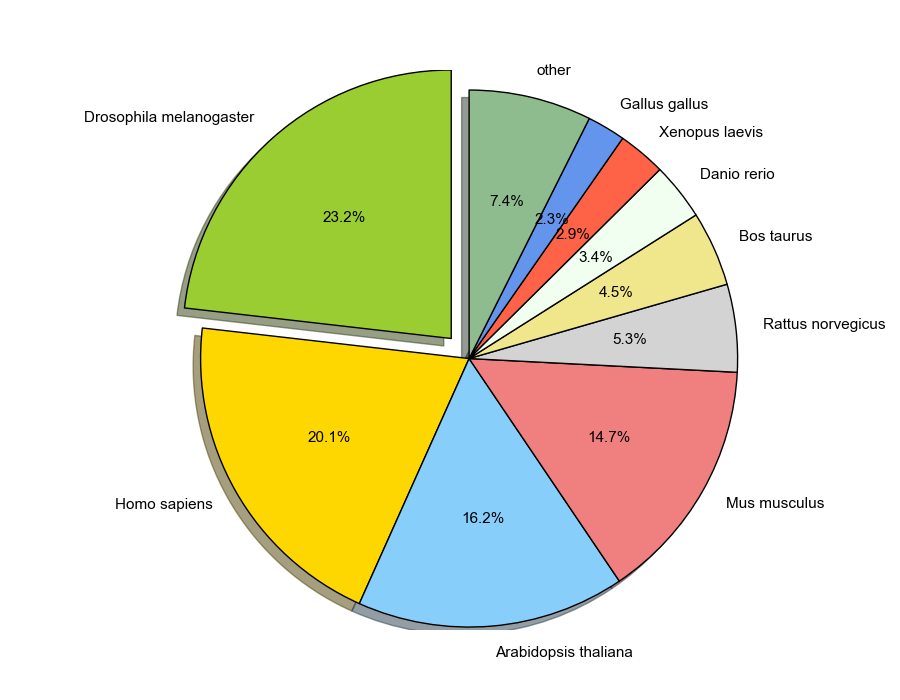
BLAST agains all known D. melanogaster proteins¶
~$ wget ftp://ftp.ensemblgenomes.org/pub/metazoa/release-28/fasta/drosophila_melanogaster/cdna/Drosophila_melanogaster.BDGP6.28.cdna.all.fa.gz ~$ gunzip -c Drosophila_melanogaster.BDGP6.28.cdna.all.fa.gz > Drome.fa ~$ makeblastdb -in Drome.fa -dbtype 'nucl' -out Drome
BLAST against all know D. plexippus¶
~$ wget ftp://ftp.ensemblgenomes.org/pub/metazoa/release-28/fasta/danaus_plexippus/cdna/Danaus_plexippus.DanPle_1.0.28.cdna.all.fa.gz ~$ gunzip -c Danaus_plexippus.DanPle_1.0.28.cdna.all.fa.gz > Danaus.fa ~$ makeblastdb -in Danaus.fa -dbtype 'nucl' -out Danaus
Summary¶
To summarize the BLAST results and map them to NCBI gene IDs.
BLAST DB Transcripts Genes Proteins SwissProt 39,457 11,285 15,584 D. melanogaster 37,342 7,963 9,672 Danaus plexippus 7,810 NA 1,392